Atypical Hemolytic Uremic Syndrome - Very Rare, Very Scary
Extremely rare, atypical hemolytic uremic syndrome is a result of a spectrum of diseases. Its potential severity makes it a significant concern for those affected.
Author:Suleman ShahReviewer:Han JuJan 19, 20241K Shares57.6K Views
Also, beyond the medical complexities, two things are clear: the chronic nature of this disease and the potential for organ damage.
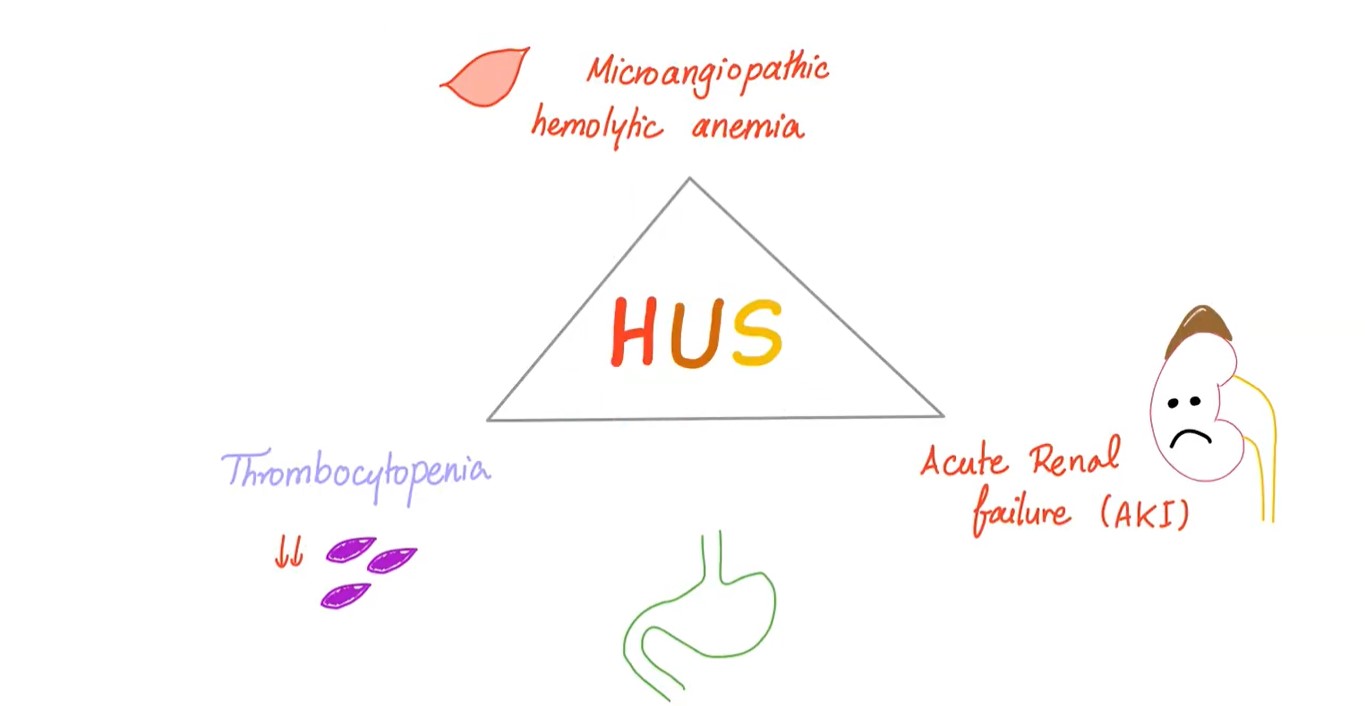
Hemolytic uremic syndrome (HUS) is defined by the association of:
- hemolytic anemia
- thrombocytopenia
- acute renal failure
Atypical hemolytic uremic syndrome stems from a variety of illnesses.
Clinical abnormalities are related with the presence of thrombotic microangiopathy (TMA).
TMA is characterized by:
- endothelial cell activation
- the release of von Willebrand factor
- platelet activation and aggregation
- leukocyte recruitment
- a procoagulant state
Escherichia coli(STEC)-HUS accounts for over 90% of cases, and usually results from infection with STEC.
Atypical hemolytic uremic syndrome accounts for 5 to 10% of cases.
It’s a heterogeneous disease.
Disorders of complement regulation are the most important reasons in the etiology of atypical hemolytic uremic syndrome.
Other causes of HUS include:
| antiphospholipid syndrome, aka Hughes syndrome | HIV (human immunodeficiency virus) |
| calcineurin-induced HUS | malignancy |
| defective ADAMTS13 function | pregnancy |
| defective cobalamin metabolism | quinine |
| genetic deficiency of thrombomodulin | systemic lupus erythematosus (SLE) |
Patients with atypical hemolytic uremic syndrome have a poor prognosis with a high mortality and morbidity in the acute phase of the disease and progression to end-stage renal disease in 50% of the cases.
We discuss aHUS and treatment options.
Pathogenesis Of Atypical Hemolytic Uremic Syndrome
Atypical hemolytic uremic syndrome is associated with defective regulation of the alternative complement pathway in over 50% of cases.
Mutations in the genes encoding the following have been reported to predispose to aHUS:
- C3
- factor B
- thrombomodulin
- autoantibodies to factor H
- complement factor I (CFI)
- complement regulator factor H (CFH)
- membrane cofactor protein (MCP: CD46)
All these changes result with over-activation of the alternative pathway.
Defective regulation of the complement activation leads to excess generation of:
- cytotoxic C5b-9
- anaphylatoxins C3a and C5a
Membrane attack complex (MAC) causes:
- cytotoxicity of endothelial cell
- intimal swelling
- cellular proliferation
This event exposes prothrombotic components in the subendothelial space and causes:
- activation of the coagulation system
- fibrin deposition
Microvascular stenosis may also result directly from:
- endothelial swelling
- subendothelial expansion
Abnormal vascular permeability mediated by C3a and C5a may cause interstitial edema of the brain and other vital organs.
The following may also occur:
| abdominal pain | pancreatitis |
| cardiac arrest/dysfunction | pericardial effusion |
| diarrhea | pleural effusion |
| dyspnea | pulmonary infiltration |
| edema | renal failure |
| mental changes | seizures |
| nausea | vomiting |
C3 level may be normal or low.
Low levels of complement C3 may indicate this complement dysregulation in atypical hemolytic uremic syndrome but it is not a definite indication.
Genetic In Atypical Hemolytic Uremic Syndrome
Mutations have been identified in approximately 50-70% of patients, including:
a. Genes encoding complement regulators
- complement regulator factor H (CFH)
- complement factor I (CFI)
- complement factor H-related proteins (CFHR)
- membrane cofactor protein (MCP)
b. Complement activators
- C3
- complement factor B (CFB)
Many different mutations in complement genes have been reported, the downstream consequences of them all are:
- over-activation of the alternative complement pathway
- excessive liberation of different cleavage fragments from C3 and C5
- formation of the lytic C5b-9 complex
This leads, in turn, to:
- platelet activation
- endothelial damage
- inflammation
- systemic microangiopathic lesions
CFH is the most important fluid-phase regulator of the alternative pathway, and it does the following:
- acts on the proteolytic inactivation of C3b
- competes with factor B for C3b binding
- accelerates the decay of C3 convertase into its components
CFH also regulates complement on host surfaces.
Most CFH mutations associated with atypical hemolytic uremic syndrome are heterozygous, it has been postulated that these mutations may exert a dominant negative effect.
Deficiency of CFH related plasma proteins and autoantibody-positive form of HUS (DEAP-HUS) is another sub-group of HUS.
It is characterized by the combination of:
- an acquired factor
- a genetic factor
The acquired factor is autoantibodies to the CFH.
These antibodies develop on a genetic background, which is mostly based on a homozygous chromosomal deletion of the CFH1 and CFH3 genes.
Autoantibodies in DEAP-HUS, as well as CFH mutations in atypical hemolytic uremic syndrome that are located within the C-terminal recognition region interfere with surface binding.
Mutant CFH shows reduced or absent binding, which results in a reduced protection of cellular host surfaces and membranes.
A mutation in membrane cofactor protein (MCP: CD46) is present with low C3b-binding and cofactor activity.
MCP mutations are more frequent in children than in adults.
C3 levels in MCP-mutated patients are most often normal.
Some of the MCP-mutated patients with decreased C3 levels have another mutation responsible for the activation of the complement in the fluid phase.
Complement factor I (CFI) mutations induce a default of secretion or disrupt its cofactor activity, with altered degradation of C3b/C4b in the fluid phase and on surfaces.
Inactivation of C3b and C4b through limited proteolytic cleavage results in the prevention of the formation of the C3 and C5 convertases and thus down-regulates the alternative and classical pathway C5.
Plasma C3 level is decreased in 20-30% of patients and CFI level in one third of patients.
Mutation in complement factor B (CFB) is also demonstrated. These mutations induce an increased stability and activity of the C3 convertase.
It results with increased complement deposition on glomerular endothelial cells.
C3 is the main component of the complement cascade. It plays a central role in the activation of classical and alternative pathways.
Most C3 mutations induce a defect of the ability of C3 to bind to MCP.
Plasma C3 concentrations are low in 70-80% of patients.
Thrombomodulin induces the activation of protein C by thrombin. It also plays a role in the inactivation of C3a and C5a.
Mutations of thrombomodulin result in a loss of cofactor activity.
Various combinations of two or more mutations were demonstrated in 12% of patients with atypical hemolytic uremic syndrome.
Genetic screening is helpful but it is not necessary for treatment planning. Genetic findings may influence long-term management of patients.
Extrarenal Involvements In Atypical Hemolytic Uremic Syndrome
Various extra renal complications due to systemic TMA may occur in hemolytic uremic syndrome (HUS), including:
- neurological,
- pancreatic; and
- cardiac involvement
Neurological involvement is the most frequent extrarenal complication in HUS.
Neurological complications, which are a major cause of morbidity and mortality, affect 10 to 48% of atypical hemolytic uremic syndrome cases.
The pathophysiology of CNS dysfunction can be multifactorial, and may involve:
- multifocal TMA
- metabolic insults (uremia, hyponatremia, and hypocalcemia)
- uncontrolled hypertension
- focal toxin-mediated mechanisms
Complement activation and subsequent C5a generation are thought to play a significant role in the progression of CNS disease.
Posterior reversible encephalopathy syndrome (PRES)-related lesions, are believed to be due to vasogenic edema and are associated with posterior white matter hyperintensity (predominantly in parieto-occipital regions).
Systemic thrombotic microangiopathy in the cerebral region can show intensities in many different regions of the brain by MRI.
Both scenarios produce similar clinical manifestations, including vision loss.
One can hypothesize that the two scenarios are interrelated in the case of atypical hemolytic uremic syndrome as:
- the edema in PRES can be caused by TMA and uncontrolled complement activation
- excessive C5a would increase vascular permeability to further exacerbate edema
While MRI findings are helpful for diagnosing neurological complications and guiding treatment, it is worth noting that there may be a delay between symptom onset and development of radiological signs.
Therapy Choices In Atypical Hemolytic Uremic Syndrome
In contrast to typical HUS, atypical hemolytic uremic syndrome is characterized by frequent relapses.
Plasma therapywas considered the first-line therapy for patients during the acute episode of aHUS and should be started within 24 hours of diagnosis.
However, in clinical practice, this is not always possible.
Plasma therapy is not successful in all patients. Patients become dependent or resistant to plasma therapy.
The necessity of life-long treatment in patients who are dependent on plasma therapy could result with reactions to plasma; worsen school performance and social life.
The morbidity and mortality of catheter placement and plasma exchange are also important issues and most studies suggest that plasma therapy often fails to rescue kidney function.
Recently in patients who are resistant to plasma therapy combined kidney-liver transplantation, a high-risk procedure was the last alternative. Combined kidney-liver transplantation should not be performed unless a patient is at high risk for life-threatening complications.
It has recently been shown that eculizumab is also an effective therapy in atypical hemolytic uremic syndrome.
Eculizumab is a humanized monoclonal anti-C5 antibody.
It blocks the alternative complement pathway at the level of proinflammatory C5a and lytic C5b-9 complex generation.
Recent case reports have shown that eculizumab may be beneficial in the long-term treatment of atypical hemolytic uremic syndrome.
Eculizumab has a high potential in this area.
The use of eculizumab over the long term has an extremely high cost, and the time of interruption is unpredictable, but it is a rescue therapy for atypical hemolytic uremic syndrome.
Also, eculizumab has a successful effect on the recovery of the renal function.
In a study published in 2013 by The New England Journal of Medicine, the authors, with Dr. Christophe M. Legendre as lead author, reported that five of the seven plasmapheresis resistant dialysis patients became free of dialysis.
Eculizumab should be administered at the recommended dosing interval.
In all atypical hemolytic uremic syndrome patients, an eculizumab serum concentration of ~50–100 μg/mL is required to provide complete inhibition of terminal complement activity.
Eculizumab should be considered for all patients without waiting for results from complement investigations, although screening for anti-CFH antibodies should be done rapidly as positive results would indicate a switch to plasma exchange and immunosuppressive drugs.
Screening for genetic complement abnormalities is needed for individualized management.
Atypical hemolytic uremic syndrome patients receiving eculizumab therapy should be monitored for TMA by measuring:
- platelet counts
- serum lactate dehydrogenase
- serum creatinine
They may require dose adjustment within the recommended 14 ± 2-day dosing schedule during the maintenance phase.
Bacterial infections should be treated promptly according to local treatment guidelines.
Vaccination may not be sufficient to prevent meningococcal infection.
All patients should be monitored for early signs of meningococcal infection, evaluated immediately if infection is suspected and treated with appropriate antibiotics if necessary.
Severe TMA complications have been observed in atypical hemolytic uremic syndrome patients after eculizumab discontinuation.
The issue of the optimal duration of eculizumab treatment has not yet been exactly known.
Treatments could be individualized on the basis of complement genetics.
Life-long treatment may be necessary in patients who have mutations associated with poor outcomes.
In the absence of other contraindications, patients with aHUS-related end-stage renal disease should be considered as eligible for renal transplantation after a thorough genetic-based assessment of their risk of recurrence.
Kidney transplantation from a living-related donor can be considered with extreme caution in rare situations but remains inadvisable if:
- the donor shares a genetic susceptibility factor with the recipient; or
- no mutations have been identified in complement genes
Prophylactic eculizumab therapy should be recommended in patients with a high risk of post-transplantation atypical hemolytic uremic syndrome recurrence and should be initiated prior to surgery at day deceased donor transplantation and include an additional dose at day one.
After these administrations, eculizumab must continue weekly.
Conclusion
Atypical hemolytic uremic syndrome arises due to a range of different diseases.
Disorders of complement regulation are the most important reasons in the etiology of aHUS.
Related to increase of experiences, eculizumab therapy may be the first-line treatment of aHUS.
We do not know the optimal duration of eculizumab therapy. We also do not know in which patient a severe relapse could be developed.
At this moment, we can suggest that in both cases as well as when it comes to atypical hemolytic uremic syndrome, eculizumab is life-saving and can enhance the quality of life.

Suleman Shah
Author
Suleman Shah is a researcher and freelance writer. As a researcher, he has worked with MNS University of Agriculture, Multan (Pakistan) and Texas A & M University (USA). He regularly writes science articles and blogs for science news website immersse.com and open access publishers OA Publishing London and Scientific Times. He loves to keep himself updated on scientific developments and convert these developments into everyday language to update the readers about the developments in the scientific era. His primary research focus is Plant sciences, and he contributed to this field by publishing his research in scientific journals and presenting his work at many Conferences.
Shah graduated from the University of Agriculture Faisalabad (Pakistan) and started his professional carrier with Jaffer Agro Services and later with the Agriculture Department of the Government of Pakistan. His research interest compelled and attracted him to proceed with his carrier in Plant sciences research. So, he started his Ph.D. in Soil Science at MNS University of Agriculture Multan (Pakistan). Later, he started working as a visiting scholar with Texas A&M University (USA).
Shah’s experience with big Open Excess publishers like Springers, Frontiers, MDPI, etc., testified to his belief in Open Access as a barrier-removing mechanism between researchers and the readers of their research. Shah believes that Open Access is revolutionizing the publication process and benefitting research in all fields.

Han Ju
Reviewer
Hello! I'm Han Ju, the heart behind World Wide Journals. My life is a unique tapestry woven from the threads of news, spirituality, and science, enriched by melodies from my guitar. Raised amidst tales of the ancient and the arcane, I developed a keen eye for the stories that truly matter. Through my work, I seek to bridge the seen with the unseen, marrying the rigor of science with the depth of spirituality.
Each article at World Wide Journals is a piece of this ongoing quest, blending analysis with personal reflection. Whether exploring quantum frontiers or strumming chords under the stars, my aim is to inspire and provoke thought, inviting you into a world where every discovery is a note in the grand symphony of existence.
Welcome aboard this journey of insight and exploration, where curiosity leads and music guides.
Latest Articles
Popular Articles