Acute Ischemic Stroke - Matrix Metalloproteinases And Blood-Brain Barrier Disruption
Acute ischemic stroke is the most prevalent kind of stroke and is caused by the abrupt obstruction of a blood artery by a thrombus or embolus, resulting in an almost instantaneous loss of oxygen and glucose to the brain region. Although numerous processes are involved in the pathophysiology of stroke, mounting evidence indicates that ischemia damage and inflammation are responsible for its pathological development.
Author:Suleman ShahReviewer:Han JuJul 10, 202279 Shares1.5K Views

Acute ischemic strokeis the most prevalent kind of stroke and is caused by the abrupt obstruction of a blood artery by a thrombus or embolus, resulting in an almost instantaneous loss of oxygen and glucose to the brain region.
Although numerous processes are involved in the pathophysiology of stroke, mounting evidence indicates that ischemia damage and inflammationare responsible for its pathological development.
Cerebral ischemia triggers cascades of pathogenic events, such as vasogenic edema, blood-brain barrier (BBB) breakdown, intracranial hemorrhage (ICH), astroglial activation, and neuronal death.
Within minutes after the commencement, this produces irreparable neuronal damage in the ischemic core.
Despite breakthroughs in the knowledge of the pathogenesis of cerebral ischemia, acute ischemic stroke therapeutic choices remain very restricted.
What Is Acute Ischemic Stroke?
When the brain's blood supply is cut off, the outcome is cellular damage that might lead to death. This is known as an acute ischemic stroke.
Sudden onset numbness or weakness in an arm and leg, facial drooping, difficulty in speaking or interpreting speech and balance or coordination difficulties are all possible stroke symptoms. A transient ischemic attack (TIA), a brief period of brain dysfunction caused by reduced blood flow, may occur before an acute ischemic stroke.
High blood pressure is the most common cause of an acute ischemic stroke. Stroke risk is further raised by a historyof transient ischemic attacks (TIAs), smoking, high cholesterol, diabetes, obesity, and atrial fibrillation, a heart rhythm issue.

What Happens During an Ischemic Stroke
Structural Components Of The Blood-Brain Barrier (BBB)
The BBB connects the peripheral and central circulatory systems. It regulates the inflow and outflow of biochemicals essential for brain metabolism and neuronal activity. The BBB's structural and functional integrity is crucial for brain homeostasis.
The anatomical substrate of the BBB is the cerebral microvascular endothelium, which forms a "neurovascular unit" necessary for CNS healthand function with astrocytes, pericytes, neurons, and ECM. Neurovascular cell–cell interactions underpin brain function. Neurovascular dysfunction causes illness. Microvessel integrity changes may impact neuronal function in the neurovascular unit. Stroke response mechanisms aren't well known. Effective stroke treatment must avoid cell death and restore neurovascular function.
Tight junctions (TJs) and adherens junctions (AJs) make up microcapillary endothelium. TJs and AJs reduce endothelial permeability. TJs are continuous membrane threads between brain ECs that comprise transmembrane proteins (JAM-1, claudins, and occludin) and cytoplasmic proteins (zonula occludens-1 to 3). Claudin-5 is a key BBB TJ adhesion molecule. Alternating TJ expression and/or distribution at the BBB during ischemic stroke may modulate their shape and function. Transmembrane and accessory proteins at the TJ are phosphorylated. Disease or medications may disrupt BBB TJ, impairing BBB function and CNS function. Understanding how BBB TJ might be damaged can help prevent and cure neurological disorders.
Matrix Metalloproteinases
Matrix metalloproteinases, also termed matrixins, break protein substrates by activating a site-bound water molecule with Zn2+. Humans have 23 MMPs (24 in mice). Excreted MMPs are classified by their substrate specificity into four classes: collagenases (MMP-1, 8, and 13), gelatinases (MMP-2 and 9), stromelysins (MMP-3, 10, and 11), and a heterogenous group containing matrilysin (MMP-7), metallo-elastase (MMP-12), enamelysin (MMP-20), endometriose (MMP-26), and epi (MMP-28). Domain structure is another way to classify MMPs. Most MMPs are latent enzymes. Activators like plasminogen activator or furin transform prohormones to active enzymes. MMPs have physically identical catalytic domains, but vary in substrate selectivity, cellular and tissue localization, membrane binding, and regulation, making them a diverse family of enzymes with numerous unknown physiological activities. All MMP family members have been associated to illness, including cancer metastasis, chronic inflammation, and neurological diseases.
The Role In BBB Disruption
Long considered biphasic, BBB disruption follows localized cerebral ischemia/reperfusion damage. Morphologically, BBB opening coincides with TJ and AJ protein redistribution from the plasma membrane to the cytoplasm and endothelial actin cytoskeleton remodelling. Ischemic insult type, intensity, and duration affect BBB disruption. Several MMPs may control BBB permeability and function during ischemic stroke.
Several MMPs are increased and activated after ischemic stroke, according to clinical and experimental investigations. MMPs degrade TJ and basal lamina proteins, causing BBB permeability, leukocyte infiltration, brain edema, and bleeding. MMP-2 and MMP-9 have diverse roles in ischemic stroke BBB breakdown. MMP-2 KO doesn't protect mice against permanent or transient MCAO. In vitro results suggest MMP-2 isn't harmful to hippocampus neurons.
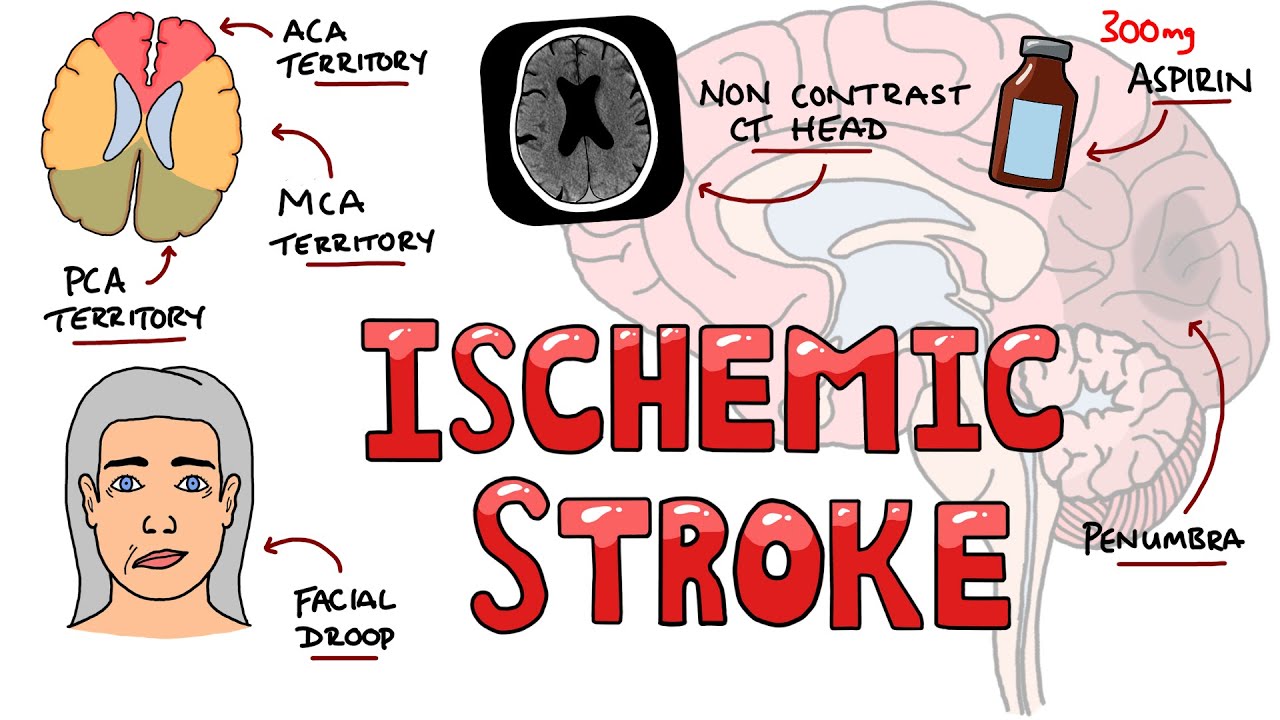
Acute Ischemic Stroke - Signs and Symptoms (Stroke Syndromes) | Causes & Mechanisms | Treatment
MMP-3 (Stromelysin-1)
Matrix metalloproteinase-3 (stromelysin-1) was identified as a 51-kDa protein in rabbit fibroblasts in 1985. MMP-3 can't destroy type I collagen, unlike collagenases. MMP-3 degrades fibronectin, denatured collagens (gelatin), laminin, and proteoglycans.
Matrix metalloproteinase-3 seems to be involved in ICH generated by tPA in mice. Suzuki et al. utilized the MCAO model to show that lack of the stromelysin-1 (MMP-3) gene in mice lowered the likelihood of ICH produced by tPA. The broad-spectrum MMP inhibitor GM6001 decreased ICH in WT but not in MMP-3-deficient mice. tPA therapyenhanced MMP-3 expression in ischemia-damaged ECs in a rat stroke model. MMP-3 may be implicated in blood vessel deterioration and ICH. MMP-9 expression was similarly enhanced in ischemic brain regions, although the quantity and distribution were similar in tPA-treated and untreated animals.
MMP-9 (Gelatinase B)
The 92-kDa proenzyme of matrix metalloproteinase-9 (gelatinase B), initially identified in neutrophils in 1974, may be activated to the mature 83-kDa enzyme. MMP-9 is the MMP that has been investigated the most in relation to acute ischemic stroke. After strokes in both human brain tissue and serum, as well as in animal stroke models starting 12 h after irreversible MCAO, MMP-9 activity is noticeably increased. High plasma MMP-9 levels are thought to be an independent predictor of HT in all stroke subtypes during the acute phase of a cerebral infarction. Claudin-5, occludin, and ZO-1 are TJ proteins that have been demonstrated to be degraded by MMP-9 in cultured brain ECs and in animal models of localized cerebral ischemia. Not only do TJ proteins be destroyed by abnormal MMP-9 proteolytic activity, but also basal membrane proteins (e.g., fibronectin, laminin, collagen, and others). In both animal models and stroke patients, this deterioration is linked to a rise in BBB permeability, which causes brain infarction, edema, and HT.
MMPs And TPA-Induced Reperfusion Injury
If started during the first three to four hours after the beginning of symptoms, thrombolysis is useful for individuals with acute ischemic stroke. The afflicted ischemic zone should be saved within this window of opportunity by thrombolysis of the blocked artery, which should also enhance the clinical prognosis. But since delayed tPA affects the cerebrovasculature, it actually raises the danger of BBB integrity disruption. The neurovascular unit may then be affected by thrombolytic tPA once it has entered the perivascular tissue. Recent research using animal models of transitory and persistent MCAO show that genetic tPA activity regulation by neuroserpin reduces BBB rupture, edema, neuronal death, and improves the prognosis of strokes. Additionally, analysis of both tPA KO and WT mice shows that endogenous tPA is both required and sufficient to cause opening of the BBB after temporary MCAO.
Numerous studies have been conducted on the mechanism of vascular unit breakdown during ischemia and tPA reperfusion. The activation of platelet derived growth factor (PDGF)-CC, a recently identified PDGF variant, may contribute to the disruption in part. Through the well-known PDGF-CC agonist, tPA increases BBB permeability. Mice were given the PDGF-α inhibitor imatinib mesylate, commonly known as Gleevec or STI571, 1 hour after MCAO to see whether the activation of PDGF-α modulates the cerebrovascular response to stroke. Comparing imatinib-treated animals to control mice, the extravasation of Evans-Blue following MCAO was reduced by 33% in the imatinib-treated animals. Additionally, imatinib, when given 1 hour after the start of ischemia, dramatically decreased hemorrhagic consequences. Treatment with PDGF-CC neutralizing antibodies decreased Evans-Blue extravasation as well.

Oxidative Stress And MMPs
In the pathogenesis of BBB degradation and ischemic stroke, oxidative stress and MMPs have a significant relationship. When the physiological balance between oxidants and antioxidants tips in favor of the oxidants, an organism is more likely to sustain possible harm. This situation is known as oxidative stress. Soon after vascular occlusion, free radicals, such as ROS and reactive nitrogen species (RNS), are produced. Accumulated free radicals not only make brain tissue more vulnerable to ischemia injury, but they also set off a variety of biochemical pathways that control the activation of MMPs. In the ischemic brain, this causes TJs to deteriorate and increases BBB permeability.
RNS and tyrosine nitration levels were shown to be elevated in the brain tissue of stroke patients as well as in experimental ischemia animal models. Additionally, in cerebral I/R damage, superoxide scavengers, NOS inhibitors, and catalysts for peroxynitrite breakdown mitigated BBB disruption, decreased infarction volume, and alleviated neurological dysfunctions. The primary defense against the toxicity of free radicals is superoxide dismutase (SOD2), which causes SOD2 KO mice to show a substantial rise in MMP-9 and a greater incidence of brain hemorrhaging following MCAO. This suggests that extra radicals have the ability to activate MMP-9 and cause HT in the post-ischemic brain. These results imply that an important therapeutic target for improving the prognosis of an ischemic stroke is free radicals.
People Also Ask
Is Acute Ischemic Stroke Life Threatening?
When symptoms of a stroke first develop, it is crucial to seek medical attention right once since they may be fatal. The signs and symptoms of an ischemic stroke often appear on one side of the body first. The American Stroke Association (ASA) advises individuals to keep F.A.S.T. in mind.
What Causes Acute Ischemic Stroke?
Ischemic strokes happen when the blood supply to a portion of the brain is cut off. The vast majority of strokes are caused by this kind of stroke. An ischemic stroke might have stopped blood flow due to a blood clot or atherosclerosis, a condition that gradually narrows the arteries.
How Is Acute Ischemic Stroke Treated?
The most effective very early therapies for ischemic stroke include: Thrombolytic treatment is administering alteplase (commonly known as tPA, or "tissue plasminogen activator") or a related drug called tenecteplase intravenously (IV) (through a vein).
What Is Considered An Acute Ischemic Stroke?
When a clot, a clump of thickened blood, blocks blood flow through a brain artery, an acute ischemic stroke takes place. According to where in the body they form, clots are either thrombotic or embolic. The more frequent of the two strokes, thrombotic stroke, happens when a clot develops in a brain artery.
Conclusion
A crucial pathophysiological event in acute ischemic stroke that begins early enough to fall inside the thrombolytic time window is blood-brain barrier disruption.
Being closely linked to HT, this early ischemic BBB damage is becoming a prospective target for lowering the hemorrhagic side effects of thrombolytic stroke treatment.
However, little is known about the processes that cause early ischemia BBB degradation. Understanding the precise function of MMPs and its signal cascades after an ischemic stroke will have significant diagnostic implications for stroke as well as for the creation of MMP-modulating treatment techniques.
Acute ischemic stroke remains one of the most difficult diseases in translational neurology due to its complex pathophysiology, which affects blood vessels, neurons, and glial cells.
Sadly, this is due to the rapid translation of experimental discoveries into clinical practice having previously raised issues. The improvements in the design of our scientific and clinical trials, together with the gains in our knowledge of the fundamental biology of acute ischemic stroke, should enable causative treatment solutions to quickly enter the clinic.

Suleman Shah
Author
Suleman Shah is a researcher and freelance writer. As a researcher, he has worked with MNS University of Agriculture, Multan (Pakistan) and Texas A & M University (USA). He regularly writes science articles and blogs for science news website immersse.com and open access publishers OA Publishing London and Scientific Times. He loves to keep himself updated on scientific developments and convert these developments into everyday language to update the readers about the developments in the scientific era. His primary research focus is Plant sciences, and he contributed to this field by publishing his research in scientific journals and presenting his work at many Conferences.
Shah graduated from the University of Agriculture Faisalabad (Pakistan) and started his professional carrier with Jaffer Agro Services and later with the Agriculture Department of the Government of Pakistan. His research interest compelled and attracted him to proceed with his carrier in Plant sciences research. So, he started his Ph.D. in Soil Science at MNS University of Agriculture Multan (Pakistan). Later, he started working as a visiting scholar with Texas A&M University (USA).
Shah’s experience with big Open Excess publishers like Springers, Frontiers, MDPI, etc., testified to his belief in Open Access as a barrier-removing mechanism between researchers and the readers of their research. Shah believes that Open Access is revolutionizing the publication process and benefitting research in all fields.

Han Ju
Reviewer
Hello! I'm Han Ju, the heart behind World Wide Journals. My life is a unique tapestry woven from the threads of news, spirituality, and science, enriched by melodies from my guitar. Raised amidst tales of the ancient and the arcane, I developed a keen eye for the stories that truly matter. Through my work, I seek to bridge the seen with the unseen, marrying the rigor of science with the depth of spirituality.
Each article at World Wide Journals is a piece of this ongoing quest, blending analysis with personal reflection. Whether exploring quantum frontiers or strumming chords under the stars, my aim is to inspire and provoke thought, inviting you into a world where every discovery is a note in the grand symphony of existence.
Welcome aboard this journey of insight and exploration, where curiosity leads and music guides.
Latest Articles
Popular Articles