Macrophages As Targets Of Developmental Immunotoxicity - A Mini-Review
Studies about macrophages as targets of developmental immunotoxicity reveal a wealth of information. For one, numerous perinatal environmental factors target macrophages.
Author:Suleman ShahReviewer:Han JuJan 19, 20242.3K Shares74.7K Views

This mini-review is among the first to focus solely on macrophages as targets of developmental immunotoxicity.
Research on disruption of immune development leading to adverse later-lifeoutcomes dates back to at least the 1970s and has fallen under the rubric of developmental immunotoxicity (DIT).
Agents of immune disruption include:
- environmental chemicals
- drugs
- diet
- physical factors (e.g. ultraviolet or UV radiation)
- psychological factors
DIT is an important component in the broader landscape of what is known as the developmental origins of adult healthand disease.
This idea of early life programming of myriad later-life diseases evolved from the more narrowly cast “Barker hypothesis,” which focused on early-life nutritional risk factors of cardiovascular diseaseoutcomes.
In the case of DIT, early insult can take two somewhat different forms.
Real-time disruption of critical maturational events can occur such as interference with positive and negative thymic selection in the thymus.
This would be likely to increase the risk of later autoimmune reactions.
Additionally, early environmental exposures can produce epigenetic alterations that result in later-life immune dysfunctional phenotypes.
These two routes to DIT may well co-exist following certain environmental exposures.
While there are a myriad of potential outcomes resulting from DIT, the most prevalent ones, based on the prevalence of immune dysfunction-related human disease, concern risk of non-communicable diseases (NCDs).
Involving virtually every tissue and organ of the body, among these conditions are:
- autoimmune
- allergic
- inflammatory
Examples of some childhood-onset conditions that have been connected to DIT include:
- asthma
- allergic rhinitis
- food allergies
- celiac disease
- type 1 diabetes
Even the later-life onset atherosclerosis appears to have early life roots given that biomarkers of atherosclerosis are evident during childhood.
Misregulated, unresolved inflammationhas been suggested as a major factor in risk of immune dysfunction-driven NCDs.
Lympho-Centric Focus Of Most DIT Studies And Assessments
While a significant number of studies and evaluation have examined developmental immunotoxicity (DIT), considerably more focus has been placed on these two than with innate immune dysfunction:
- adaptive immune responses
- lymphoid cell surface markers as indicators of DIT
In fact, the most widely used functional assessment for immunotoxicity screening in general including DIT has been the T-dependent antibody assays (TDAR), often measuring IgM response to sheep erythrocytes or keyhole limpet hemocyanin (KLH) as the endpoint.
The argument that has been made is that antigen presenting cell (APC) activity is included in the TDAR measure; hence, professional APCs such as macrophages do have some input.
But in reality, the key measurements pertaining to risk of immune dysfunction producing NCDs are more likely to fall among macrophages and other innate immune cells responding to challenges in tissues.
Given the concern over misregulated inflammation associated with NCDs and potential targeting of tissue macrophages (e.g. microglia, alveolar macrophages, and Kupffer cells) by environmental agents, the present mini-review focuses solely on the macrophage as a target of DIT.
There are many biomarkers and assessment tools available for the evaluation of macrophage cell populations and function.
A recent review discussed the assays that can be applied to an examination of immunotoxicity involving macrophage.
Macrophages - Distribution And Diversity
Recognition of their many functions has evolved from an original view as a primary vehicle for removing dead and dying cells to a far more global role as a homeostatic regulator of tissues.
In fact, because macrophages can take on such a diverse range of form and function and have been given other names connected with their resident tissues during much earlier immunologyand cell biology research, their cell-lineage origins had been unclear.
The picture below shows the many varied forms of macrophages as distributed in tissues:
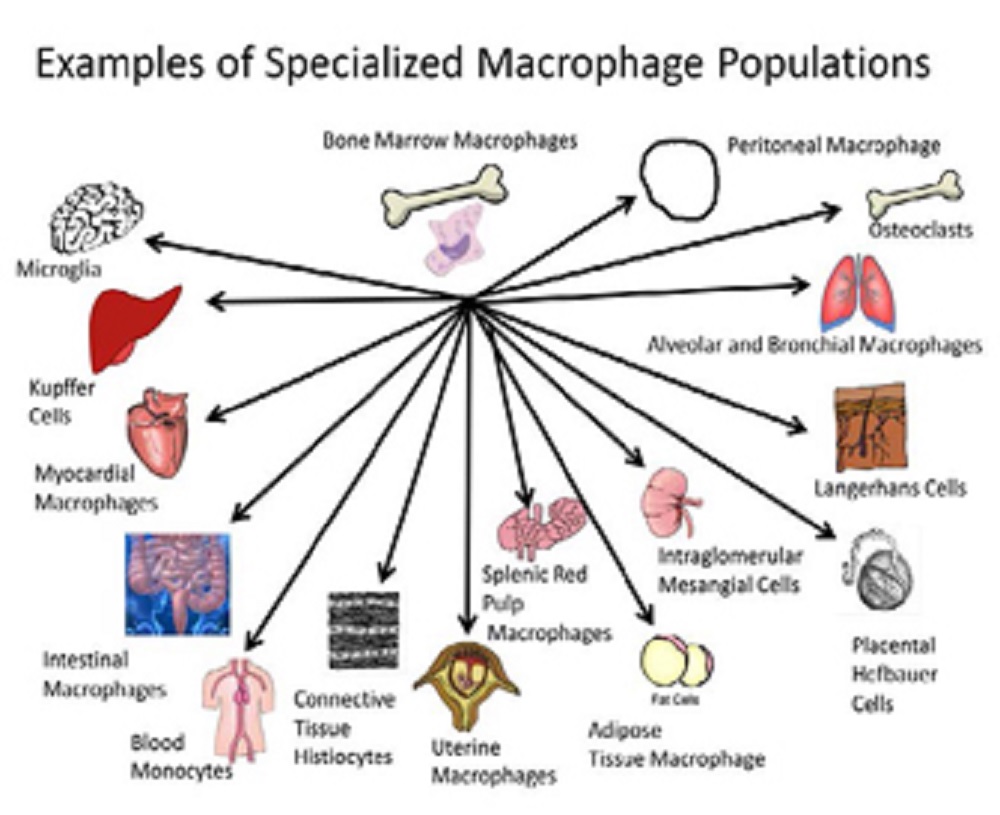
Macrophages populate most tissues and organs of the body performing both highly specialized functions and regulating organ homeostasis.
Regarding the picture, examples of different macrophage populations associated with different tissues are illustrated. Each population may have its own sensitivity and profile for environmentally-induced developmental immunotoxicity.
From the standpoint of toxicant-induced alteration of function, each of these specialized forms may have its own characteristics and sensitivities related to environmental insult.
For example, mineral fibers are thought to have a higher persistence rate in the peritoneal cavity versus the lung based on differences in peritoneal versus alveolar macrophage phagocytic activity and subsequent responses.
Macrophages serve as an anchor of innate immune function via:
- molecular pattern recognition
- phagocytic
- chemokine recruitment of cells
- cytokine orchestration
- metabolic control of the local environment
- hormonal modification of surrounding cells
- tumor
- virus-infected killing functions
In addition to participating in antigen presentation for adaptive immunity, macrophages also provide an important homeostatic function in virtually all tissues and organs.
This includes not only the removal of dead cells and repair processes within tissues but also the sampling at portals of the exposure to the external environment, such as:
- airways
- gastrointestinal tract
- urogenital tract
Macrophages sense microbes, allowing them to interact with both the:
- normal commensal microbiota
- invading pathogens
They are also a front line in detection as well as in the handling and/or transport of potentially harmful environmental contaminants (e.g., airborne particulate matter).
Given their locations in organs and tissues, range of critical host-wellness functions, and interactions with the environment, it is not surprising that their status can often dictate organ and tissue status.
Their specific distribution and presence in so many tissues means that they are invariably among the cells that first receive significant environmental exposures.
Whether it is inhaled diesel exhaust particles or mycobacteria, UVB skin radiation or oral exposure to plasticizers, resident macrophages are at the portal of environmental exposure and are among the first cells to handle the chemical or drug.
In fact, the manner in which they first interact with and handle the environmental risk factors can often set the template for the longer-term health risks for the host.
Macrophages - Plasticity And Polarization
In responding to challenges to the host, macrophages can shift among a variety of phenotypes to participate in different overlapping phases of first inflammation followed by:
- cell proliferation
- angiogenesis
- matrix remodeling
In her study published in 2013 by the journal Inflammation Research, Selma Giorgio argued that macrophages are potentially the cell type with the most phenotypic flexibility and that the balance of shifting among macrophage phenotypes (such as M1 vs. M2 macrophages) is a response to environmental signals.
The picture below shows examples of environmental factors that can promote either M1 or M2 polarization:
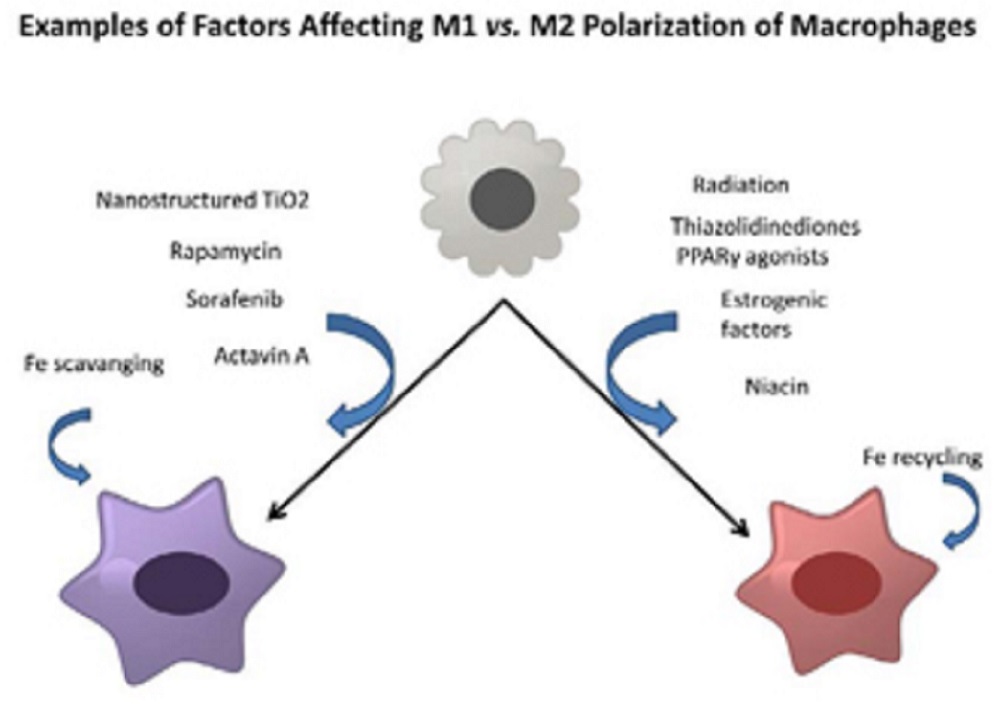
The polarization of macrophages between M1 versus M2 cells exerting a significant influence over the host defense and repair activities partitioning antimicrobial, pro-inflammatory activities in tissues versus anti-inflammatory, repair and growth-promotion functions that can include even the growth promotion of tumors.
Several different classes of environmental factors have been identified that affect this polarization. These include:
- drugs
- metals
- estrogen-endocrine status
The latter suggests that many endocrine disrupting chemicals may similarly affect this pathway.
Macrophage populations can polarize into largely pro-inflammatory cells (M1) capable of carrying out not only antimicrobial functions but also tissue destruction or, alternatively, the polarization can skew towards growth-promoting cell populations (M2) involved in:
- repair
- cell proliferation
- healing
M2 macrophages have also been involved with tumor cell proliferation.
Subgroups of M2 macrophages have also been defined based on:
- induction signals
- cell surface markers
- functional distinctions
The extent to which environmental factors can alter the capacity for or balance of macrophage polarization in response to host challenge can greatly affect the risk of specific pathological outcomes.
Dysfunction among macrophages can vary widely resulting in such adverse outcomes as:
- fibrosis
- failure to heal
- misdirected and/or misregulated inflammation causing tissue damage, pathology, and loss of function
Ineffective antigen presentation is also a potential functional problem among macrophages.
Reported Environmental Risk Factors For DIT Of Macrophages
While developmental immunotoxicity (DIT) studies date back to the 1970s, only a subset of these studies included macrophages and/or macrophage-driven functions in the adverse outcome assessment.
Despite this limitation, a broad range of environmental risk factors for the DIT of macrophages has been reported in the literature.
It is apparent that the same categories of risk factors producing DIT also target macrophages.
Few studies have compared different macrophage populations for differential sensitivity.
Since most studies are connected to a specific disease model, analyses are often limited to one specialized population of macrophages located in the target tissue of interest, such as:
- alveolar macrophages
- testicular macrophages
- microglia
- Kupffer cells
Environmental risk factors include:
a. Metals
- lead
- cadmium
- arsenic
- manganese
b. Endocrine disrupting chemicals
- bisphenol A
- di-2-ethylhexyl phthalate (DEHP)
Other factors also include:
- pesticides (chlordane)
- air pollutants
- dietary factors
- maternal stressors
- drugs
- alcohol
- nicotine
- combustion products
- volatile organic compounds (e.g., toluene)
- physical factors (e.g., UVB radiation)
- component of infectious agents (LPS)
- nonylphenol
- 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin
Some combinations of exposure were also found to produce macrophage toxicity (e.g., diesel exhaust exposure combined with maternal stress or juvenile high-fat diet).
As expected, reported alterations of DIT in macrophages cover a wide range of changes, including:
- changes in cell number
- reduced phagocytic activity
- altered toll-like receptor (TLR) expression and/or signaling
- skewed cytokine production (often favoring pro-inflammatory cytokine production)
- altered metabolite production
- altered cytotoxic activity towards tumor cells or microbes
Disrupted Macrophage Homeostasis And Non-Communicable Diseases
Macrophages appear to play a pivotal role in the promotion of NCDs that comprise a wide range of tissues.
Therefore, these things increase the likelihood of later tissue-focused pathology and disease:
- disruption of normal macrophage seeding of tissues
- maturation
- acquisition of functional capacities related to tissue homeostasis
Examples of the involvement of dysfunctional macrophage populations with NCDs include:
- atherosclerosis
- other cardiovascular diseases
- type 2 diabetes
- non-alcoholic hepatic steatosis
- asthma
- chronic obstructive pulmonary disease
- obesity
- Alzheimer’s disease
Critical Windows Of Macrophage Vulnerability
As with other components of the developing immune system, macrophages in the non-adult appear to be more sensitive to environmental insult and functional alteration than those of the adult.
This increased sensitivity of the young (vs. adults) is not restricted only to prenatal development but also includes postnatal (e.g. juvenile) periods of development.
For at least one toxicant, macrophages appear to have a broader window of increased sensitivity compared with certain lymphoid populations.
In a study published in 2001 by the journal Toxicological Sciences, the authors, with Terry L. Bunn as lead author, found that:
- prenatal lead exposure in rats across early and late windows of gestation produced persistent macrophage toxicity; whereas,
- lead-induced T-helper cell alterations were restricted to exposures during later embryonic development
However, there is a dearth of information on distinctions among narrower developmental windows within prenatal, adolescent, or juvenile periods.
The picture below shows windows of macrophage sensitivity described for various environmental risk factors from a selection of studies:
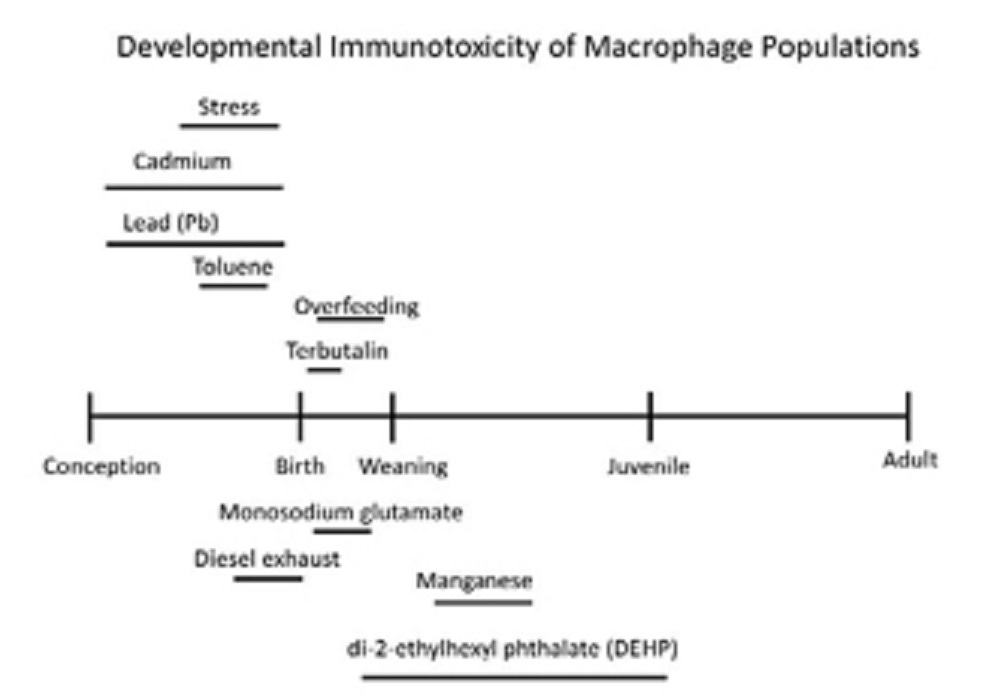
It reinforces the importance of both prenatal and postnatal developmental windows during which specific macrophage populations may possess enhanced sensitivity for DIT.
Conclusion
Macrophages are a ready target of developmental immunotoxicity (DIT), and the ramifications of adverse macrophage outcomes of DIT usually result in an increased risk of NCDs.
These diseases manifest in a wide array of tissues mostly supported by misregulated inflammation.
While macrophages have not been a pivotal focus within immunotoxicology or its sub-category, DIT, there is ample evidence to suggest they deserve more attention.
In the handful of studies where comparative sensitivity in early development has been measured across the immune system and other cellular components, macrophage parameters are among the most sensitive indicators of toxicity.
An example of this is found in a study published in 2012 by the journal Toxicology and Applied Pharmacology, with Elisa C. M. Tonk as lead author, where innate immune inflammatory markers, including those from macrophages, were among the most sensitive indicators of juvenile immunotoxicity to DEHP exhibiting far greater sensitivity than several reproductive endpoints.
In the effort to reduce the prevalence of environmentally-mediated NCDs beginning in early life, more attention needs to be paid to the impact of environmental factors on different macrophage populations.
Further studies should also be made about macrophages as targets of developmental immunotoxicity.
Jump to
Lympho-Centric Focus Of Most DIT Studies And Assessments
Macrophages - Distribution And Diversity
Macrophages - Plasticity And Polarization
Reported Environmental Risk Factors For DIT Of Macrophages
Disrupted Macrophage Homeostasis And Non-Communicable Diseases
Critical Windows Of Macrophage Vulnerability
Conclusion

Suleman Shah
Author
Suleman Shah is a researcher and freelance writer. As a researcher, he has worked with MNS University of Agriculture, Multan (Pakistan) and Texas A & M University (USA). He regularly writes science articles and blogs for science news website immersse.com and open access publishers OA Publishing London and Scientific Times. He loves to keep himself updated on scientific developments and convert these developments into everyday language to update the readers about the developments in the scientific era. His primary research focus is Plant sciences, and he contributed to this field by publishing his research in scientific journals and presenting his work at many Conferences.
Shah graduated from the University of Agriculture Faisalabad (Pakistan) and started his professional carrier with Jaffer Agro Services and later with the Agriculture Department of the Government of Pakistan. His research interest compelled and attracted him to proceed with his carrier in Plant sciences research. So, he started his Ph.D. in Soil Science at MNS University of Agriculture Multan (Pakistan). Later, he started working as a visiting scholar with Texas A&M University (USA).
Shah’s experience with big Open Excess publishers like Springers, Frontiers, MDPI, etc., testified to his belief in Open Access as a barrier-removing mechanism between researchers and the readers of their research. Shah believes that Open Access is revolutionizing the publication process and benefitting research in all fields.

Han Ju
Reviewer
Hello! I'm Han Ju, the heart behind World Wide Journals. My life is a unique tapestry woven from the threads of news, spirituality, and science, enriched by melodies from my guitar. Raised amidst tales of the ancient and the arcane, I developed a keen eye for the stories that truly matter. Through my work, I seek to bridge the seen with the unseen, marrying the rigor of science with the depth of spirituality.
Each article at World Wide Journals is a piece of this ongoing quest, blending analysis with personal reflection. Whether exploring quantum frontiers or strumming chords under the stars, my aim is to inspire and provoke thought, inviting you into a world where every discovery is a note in the grand symphony of existence.
Welcome aboard this journey of insight and exploration, where curiosity leads and music guides.
Latest Articles
Popular Articles