The Interleukin-23/Interleukin-17 Axis And The Role Of Treg/Th17 Cells In Rheumatoid Arthritis And Joint Destruction
Th17 cells contribute directly to bone loss by producing a certain kind of protein called RANKL. That is only but one part played by Th17 cells in rheumatoid arthritis.
Author:Suleman ShahReviewer:Han JuFeb 12, 2024329 Shares32.9K Views
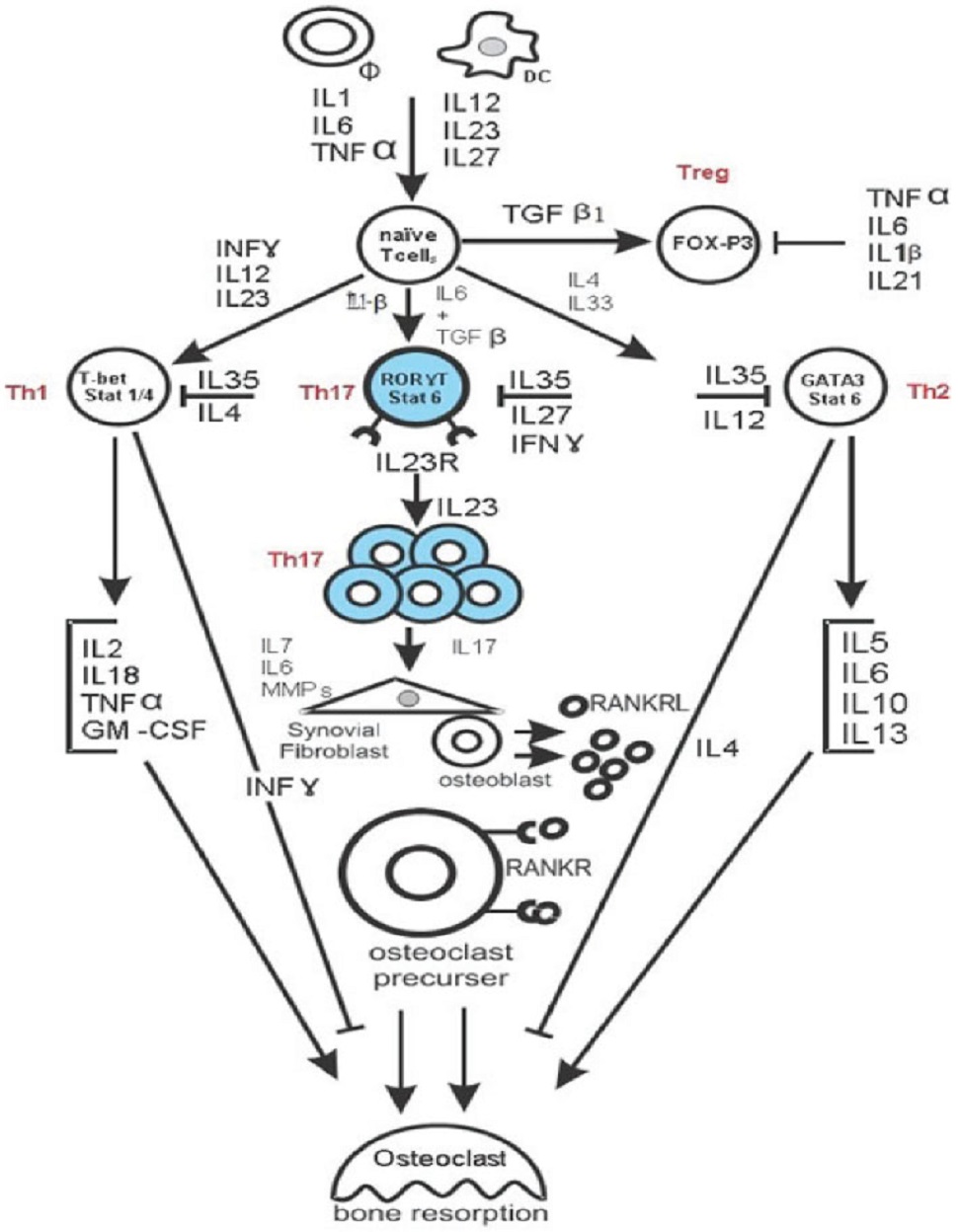
Let us discuss the role of Th17 cells in rheumatoid arthritis.
Rheumatoid arthritis is caused by a complex mechanism involving synoviocytes, osteoclasts, and immune cells that are interconnected via cytokines and other signaling molecules.
Effective medical treatment for joint destruction in rheumatoid arthritis is lacking because the molecular mechanisms leading to joint destruction are incompletely understood.
However, it is known that cytokine-mediated immunity and perturbations in the balance of effector cells at the disease site play a crucial role in rheumatoid arthritis pathogenesis.
Cellular and cytokine inhibitors have been used for treatment purposes.
Increasing evidence has revealed the importance of IL-17, an activated T cell-derived inflammatory cytokine, and IL-23 in the pathogenesis of rheumatoid arthritis.
The role of IL-17 has been of particular interest, as IL-17 affects the differentiation and activation of pathogenic osteoclasts.
Cellularly, the key features of rheumatoid arthritis include disturbance of the Th17/Treg balance and plasticity, deregulated Th17 responses and the reduction or absences of Treg cells.
Recent developments in the area of CD4[+] T-cell differentiation, together with experimental and preclinical findings on inhibitors of the IL-17 pathway and the use of Treg cell-based therapy, indicate that CD4[+] effector cells, TH17 and Treg, could be effective targets for restoring immune tolerance.
In this critical review, we summarize the progress in our understanding of the role of IL-23, IL-17 and CD4[+] T-cell differentiation into specialized effectors, focusing on Th17 and Treg cells, in the pathogenesis of rheumatoid arthritis.
Abbreviations List
Refer to this table for some of the abbreviations found in this review:
| DC | dendritic cells |
| GM-CSF | granulocyte-monocyte colony-stimulating factor |
| IFN | interferon |
| IL | interleukin |
| OPG | osteoprotegerin |
| RA | rheumatoid arthritis |
| RANKL | receptor activator for NF-κB ligand |
| TGF | tumor growth factor |
| Th17 | interleukin-17-producing helper T cells |
| TNF | tumor necrosis factor |
| Treg | T regulatory cells |
Preliminary Discussion
Rheumatoid arthritis (RA) is a chronic, systemic autoimmune disease.
In RA, inflammationusually affects multiple peripheral joints, but other tissues and organs are also involved.
RA can affect any joint that has a synovial-fluid-filled joint cavity lined by a synovial membrane and regions in which cartilage overlies the bone.
A study published in the journal Frontiers in Immunologyin 2012 suggested that the clinical progression of RA involves three stages in the progression from an immune response to bone destruction.
These are summarized as follows:
a. Initiation phase
An antigen(s) triggers immune responses, and symptoms are not yet observed in the joints.
b. Inflammatory phase
This phase begins when inflammatory symptoms, such as swelling, are recognized in the joints and continue until structural changes occur.
c. Bone and cartilage destruction phase
In this phase, structural damage is observed in the bone and cartilage.
Disease progression occurs slowly between initiation and the observation of clinical symptoms.
Although the precise mechanisms of RA pathogenesis are still unclear, it is generally well recognized that numerous inflammatory cells and the extensive production of pro-inflammatory mediators by these cells, are involved in:
- chronic joint inflammation
- progressive cartilage destruction
- osteoclast-mediated bone destruction
Some of these inflammatory cells include:
- B cells
- dendritic cells (DCs)
- fibroblast-like synoviocytes
- macrophages
- monocytes
- plasma cells
- T-cells
In simple words, RA is caused by a complex mechanism involving:
- immune cells
- osteoclasts
- synoviocytes
They communicate via cytokines and other signaling molecules.
Herein, we describe the involvement of certain cytokines and cells that play a major role in RA pathogenesis, particularly in osteoclastogenesis.
Several pro-inflammatory cytokines and the newly recognized cytokines IL-23 and IL-17, have been extensively examined for their role in the disease process in arthritis in termsof their:
- expression
- functional activity in synovial tissues
- their use as prognostic factors
Some pro-inflammatory cytokines include:
- tumor necrosis factor (TNF)-α
- interleukin (IL)-1β
- IL-6 and interferon (IFN)-γ
Some of these cytokines are now targeted in the standard treatment of RA patients, and others, such as IL-23, are being tested as targets in the clinic with promising results.
IL-23 will be one focus of this critical review.
IL-23, which is a pro-inflammatory, heterodimeric cytokine, is expressed primarily by macrophages and DCs.
Upon activation by tumor growth factor (TGF)-β and IL-6, its receptor (IL-23R) is found on:
- DCs
- memory T-cells
- naïve T-cells
- natural killer T-cells
- macrophages
IL-23 belongs to the IL-12 cytokine family and is composed of two subunits, p19 and p40; the p40 subunit is shared with IL-12.
Both IL-12 and IL-23 are involved in the expansion of activated IL-17-producing helper T (Th17) cells (Figure 1).
Th17 cells play a key role in RA pathogenesis and produce:
- IL-17A
- IL-17F
- IL-6
- IL-22
- TNF-α
- granulocyte-macrophage colony-stimulating factor (GM-CSF)
IL-12 and IL23 are expected to play separate roles at different stages of the inflammatory response. IL-12 promotes the initiation of the inflammatory response by inducing the development of Th1 cells, which produce IFN-γ, whereas IL-23 regulates late-stage inflammatory processes and is involved in the expansion of Th17 cells.
Recent evidence suggests that IL-23, rather than IL-12, is the crucial factor in the pathogenesis of various immune-mediated disorders.
IL-23 expression is important in the inflammatory response not only in RA but also in psoriasis and inflammatory bowel disease.
IL-23 is found:
- in the synovial membrane of RA patients
- in the skin of psoriasis patients
- in the bowel wall of patients with chronic inflammatory bowel disease
Inflammatory macrophages express IL-23R and are activated by IL-23 to produce IL-1, TNF-α and IL23 itself, which is a known as the autocrine loop of IL-23 in macrophages.
Moreover, an association of IL-23 and IL-23R polymorphisms, with susceptibility to RA, has been reported.
Therefore, IL-23 and its receptor are promising treatment targets for such inflammatory diseases.
Ustekinumab, which inhibits IL-12 and IL-23 by blocking p40, has been shown to be effective at treating:
- cutaneous psoriasis
- psoriatic arthritis
- Crohn’s disease
Treatments that specifically target IL-23 (antibodies specific for p19) are being developed.
This critical review discusses the IL-23/IL-17 axis and the role of regulatory T-cells (Treg) or Th17 cells in RA and joint destruction.
Main Discussion
Role Of The IL-23p19/IL-17 Axis In RA
IL-23p19 and IL-17 correspond to a new axis that drives immune activation and chronic inflammation through the differentiation and activation of Th17 cells.
Both IL-17 and IL-23 were found to be present in patients with RA in their:
- serum
- synovial fluid
- synovial tissue
They were absent in healthy joints and other types of arthritis, such as osteoarthritis.
The significant role of IL-17 in RA is highlighted by the current success of clinical trials, with neutralizing antibodies specific for IL-17.
Blocking IL-17 during the reactivation of antigen-induced arthritis diminishes:
- joint swelling
- joint inflammation
- bone erosion
This cytokine has diverse immune functions, including:
- activation of synovial fibroblasts to produce IL-6, IL-8 and vascular growth factors
- chemo-attraction of neutrophils
- stimulation of the expression of the receptor activator of NF-κB ligand (RANKL), a factor that is crucial for osteoclastogenesis
- mediation of direct pro-osteoclastogenic effects
In addition, IL-23 plays an important role in RA in animal models.
IL-23 gene-targeted mice do not develop clinical signs of arthritis and are resistant to the development of joint and bone pathology.
Moreover, this resistance correlates with an absence of IL-17-producing CD4[+] T-cells.
Other than regulating the expansion of Th17 cells, IL-23 induces IL-17 production by Th17 cells and TNF-α, IL-6, IL-22, GM-CSF and other novel factors associated with the induction of autoimmune inflammation.
The serum and synovial fluid levels of IL-23 are correlated positively with the concentration of IL-17, in addition to that of other cytokines such as TNF-α and IL-1β, indicating that IL-23 is closely linked to the production of other pro-inflammatory and anti-inflammatory cytokines in the course of RA.
These findings highlight the importance of the IL-23p19/IL-17 axis as an essential inflammatory mediator in the destructive phases of autoimmune arthritis.
Th17/RANKL Is The Bridge Between The Immune And Skeletal Systems And Leads To Bone Destruction In RA
Bone destruction in RA is mainly attributable to the abnormal activation of osteoclasts.
Osteoclasts are multinucleated cells of the monocyte/macrophage lineage that degrade bone matrix and dynamically remodel the skeleton.
The generation of osteoclasts is physiologically supported by mesenchymal cells such as osteoblasts, which provide essential signals for differentiation of the osteoclast lineage:
- macrophage colony-stimulating factor
- receptor activator of nuclear factor kappa beta (NFkB ligand) or RANKL
- costimulatory signals for RANKL
Osteoprotegerin (OPG) is a soluble decoy receptor for RANKL that blocks the pro-osteoclastogenic activity of RANKL.
In inflammatory arthritis, the RANK/RANKL pathway is activated, resulting in deregulated bone remodeling.
In patients with RA, the synovial tissues exhibit an increased ratio of RANK:OPG mRNA expression, indicating that pro-osteoclastogenic conditions dominate the microenvironment of RA-affected joints.
There is accumulating evidence that bone destruction in RA is driven by the Th17-cell-mediated induction of RANKL expression in synovial cells, which, together with inflammatory cytokine production, stimulates the differentiation and activation of osteoclasts and therefore bone resorption.
IL-17 has been shown to increase the spontaneous production of matrix metalloproteinases by synoviocytes and activate chondrocytes to inhibit matrix synthesis and induce cartilage matrix degradation.
In mice, intra-articular injection of IL-17 into the knee joint results in joint inflammation and damage.
In addition, it is well documented that blocking IL-17/IL-17R signaling is effective at controlling local and systemic inflammatory manifestations, lowering the level of RANKL and blocking cartilage and bone destruction.
These findings indicate that Th17 and RANKL bridge the skeletal and immune systems and have led to a new research field called osteoimmunology.
This interdisciplinary field is important for the treatment of diseases associated with the bone and immune systems.
Perturbations In The Balance Between Treg And Th17 Cells Lead To Osteoclastogenesis And Bone Destruction
A study published in the Journal of Experimental Medicinein 2004 raises an important question.
Is it only a reduction in the number of Treg cells that initiates RA, or is it a qualitative defect in Treg cells that induces the aggressiveness of RA?
The authors of the said study have presented evidence suggesting that Treg cells derived from patients with active RA are defective in the ability to suppress cytokine production and to convey a suppressive phenotype to CD4[+] effector T cells.
They demonstrated that the clinical response to anti–TNF-α therapy, but not conventional therapies, in RA patients, was correlated with an increased number of peripheral blood Treg cells.
One possibility is that the presence of TNF-α and other pro-inflammatory cytokines may hinder the ability of Treg cells to prevent autoimmune disease.
The role of Treg cells in RA has been reported in both patients and animal models, where the Treg subset represents approximately 5% to 10% of the CD4[+] T-cell population.
Increasing evidence suggests that Treg cell function is impaired in chronic inflammatory diseases such as RA.
Interestingly, many studies have shown that:
a. Tregs directly suppress differentiation into osteoclasts in vitro.
b. Treg depletion has been shown to aggravate various experimental autoimmune diseases, including collagen-induced arthritis.
The underlying mechanism is not fully understood; however, pro-inflammatory cytokines (e.g., TNF-α, IL-6, IL-1β, and IL-21), which are present within the synovial environment, can reduce the suppressive function of Treg cells in vitro.
Therefore, the potential for Treg cells to modulate ongoing inflammation in RA remains unclear.
The plasticity between the Th cell subsets is worth mentioning here.
It was thought for many years that all CD4[+] T-cells (Th1, Th2, Th17, and Treg cells) are terminally differentiated lineages with a stable phenotype.
However, recent evidence suggests that these cells display plasticity, as observed by the ability to change their pattern of expression of both cytokines and master regulator transcription factors in response to external stimuli, suggesting that apparently fully committed Th cells have a plasticity feature.
However, the Th17 subset of Th cells is pro-inflammatory.
It plays vital roles in host defense and has been shown to be involved in the pathogenesis of autoimmune and inflammatory diseases primarily by secreting IL-17A and other cytokines, such as IL-21 and IL-22.
It is therefore clear that Th17 and Treg cells have a functional antagonism, in which:
a. Tregs act as immunosuppressive cells; and
b. Th17 cells are involved in inducing autoimmunity.
There is also an opposition in their generation.
Both Th17 and Treg cells are induced from uncommitted CD4[+] T-cells by different cytokine-driven signaling pathways.
The IL-6/STAT3 pathway is crucial for differentiation into Th17 cells and completely inhibits the TGF-β–induced generation of Foxp3[+] Tregs.
The deletion of STAT3 in CD4[+] T cells impairs differentiation into Th17 cells and production of IL-17A and limits the development of experimental autoimmune encephalomyelitis.
By contrast, the IL-2/STAT5 pathway is essential for the maintenance of homeostasis and for the competitive fitness of Treg cells, and it suppresses differentiation into Th17 cells.
Treg cells and Th17 effectors arise in a mutually exclusive fashion, depending on whether they are activated in the presence of TGF‐β or TGF‐β plus IL-6.
In a healthy individual or in the absence of any inflammatory trigger, TGF‐β produced in the immune systemwill suppress the generation of Th17 effector cells and induce Foxp3[+] Treg cells and thereby maintain self‐tolerance.
However, during infection or inflammation, IL-6 produced by the activated innate immune system suppresses the generation of TGF‐β−induced Treg cells and induces a pro‐inflammatory T‐cell response predominated by Th17 cells.
Although IL-6 plays a critical role in the development of the Th17 response and in the inhibition of Treg functions, it is also thought that additional control steps must exist during this process and that other cytokines might participate in differentiation into Th17 cells.
Th17 cells and CD4[+]CD25[+]Foxp3[+] Treg cells are novel CD4[+]Th cell subsets that are distinct from Th1 and Th2 cells.
Treg cells are an immunosuppressive CD4[+]Th cell subset that contribute in maintaining peripheral self-tolerance and preventing the development of various inflammatory diseases through direct contact with effector immune cells and the secretion of anti-inflammatory cytokines, such as IL-10 and TGF-β1.
It has been proposed that Treg cells are generated in the thymus, presumably by a moderately high-affinity interaction between developing autoreactive T-cell receptor‐bearing T cells and self‐antigens presented in the thymus.
Once generated, Treg cells are seeded into the peripheral immune compartment, where they regulate the activation and effector functions of auto-reactive T-cells throughout the lifeof an individual.
Foxp3 is a transcription factor that is specifically expressed in CD4[+]CD25[+] Treg cells.
Its expression is crucial for the anergic phenotype of these cells in vitro and for their suppressor function.
TGF-β1 is a critical differentiation factor for the generation of Tregs.
It has been revealed that the effector function of Th cells (i.e., Th1, Th2, and Th17 cells) is regulated by Tregs.
Conclusion
In this critical review, we have summarized the progress in our understanding of the role of the cytokines IL-23 and IL-17 and the differentiation of CD4[+] T-cells into specialized effectors, focusing on Th17 and Treg cells, in the pathogenesis of RA.
We conclude that the pathogenesis of RA is slow but complex and that both cytokines and cell-mediated immunity are involved in a complex process that eventually results in irreversible bone and cartilage destruction.
Cytokine levels and the Treg/Th17 balance might be relevant to the diversity of clinical manifestations in RA patients and might be critical for the development of improved clinical management and therapeutic options.
Here are some further pointers:
a. Targeting the IL-23/Th17 axis for drug therapy appears to be a realistic approach to alternative treatments of chronic inflammatory diseases.
b. IL-23 induces osteoclastogenesis in humans via the induction of IL-17 expression.
c. RANKL, expressed on T-cells, osteoblasts, and synovial fibroblast, induces osteoclastogenesis.
d. Th17 and RANKL bridge the skeletal and immune systems and have led to a new research field called osteoimmunology.
e. IFN-γ has been reported to inhibit osteoclastogenesis.
f. CD4[+]CD25[+]Foxp3[+] Treg cells and CD3[+]CD4[+]IL-17-producing Th17 cells are two subsets of CD4[+] Th cells.
g. The Treg/Th17 balance is regarded as a key factor in immune homeostasis, and an imbalance in this ratio is associated with disease activity in several autoimmune diseases.
h. Disturbances in the number and/or function of Treg cells are a feature of autoimmunity.
This critical review strives to make significant contributions in studies related to Th17 cells in rheumatoid arthritis.

Suleman Shah
Author
Suleman Shah is a researcher and freelance writer. As a researcher, he has worked with MNS University of Agriculture, Multan (Pakistan) and Texas A & M University (USA). He regularly writes science articles and blogs for science news website immersse.com and open access publishers OA Publishing London and Scientific Times. He loves to keep himself updated on scientific developments and convert these developments into everyday language to update the readers about the developments in the scientific era. His primary research focus is Plant sciences, and he contributed to this field by publishing his research in scientific journals and presenting his work at many Conferences.
Shah graduated from the University of Agriculture Faisalabad (Pakistan) and started his professional carrier with Jaffer Agro Services and later with the Agriculture Department of the Government of Pakistan. His research interest compelled and attracted him to proceed with his carrier in Plant sciences research. So, he started his Ph.D. in Soil Science at MNS University of Agriculture Multan (Pakistan). Later, he started working as a visiting scholar with Texas A&M University (USA).
Shah’s experience with big Open Excess publishers like Springers, Frontiers, MDPI, etc., testified to his belief in Open Access as a barrier-removing mechanism between researchers and the readers of their research. Shah believes that Open Access is revolutionizing the publication process and benefitting research in all fields.

Han Ju
Reviewer
Hello! I'm Han Ju, the heart behind World Wide Journals. My life is a unique tapestry woven from the threads of news, spirituality, and science, enriched by melodies from my guitar. Raised amidst tales of the ancient and the arcane, I developed a keen eye for the stories that truly matter. Through my work, I seek to bridge the seen with the unseen, marrying the rigor of science with the depth of spirituality.
Each article at World Wide Journals is a piece of this ongoing quest, blending analysis with personal reflection. Whether exploring quantum frontiers or strumming chords under the stars, my aim is to inspire and provoke thought, inviting you into a world where every discovery is a note in the grand symphony of existence.
Welcome aboard this journey of insight and exploration, where curiosity leads and music guides.
Latest Articles
Popular Articles